Biography
I study the evolution of complex traits, such as color patterns, brains and behavior. To that end, I combine large-scale phenotypic and genetic datasets with methodological advances, such as machine learning and statistical modelling.
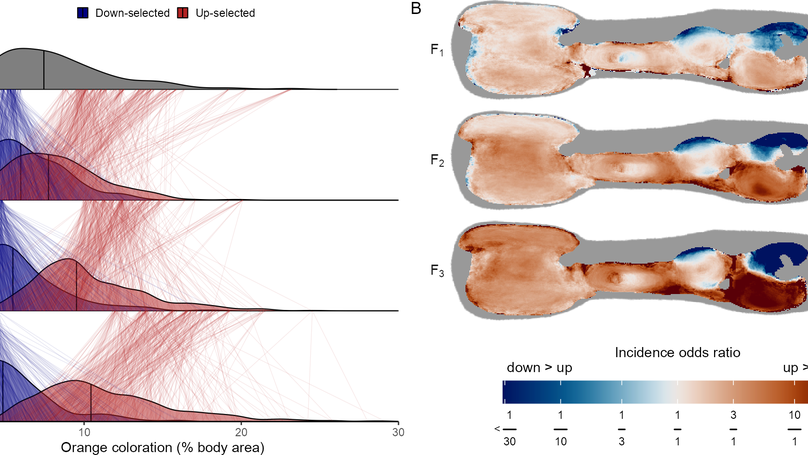
I am currently working as a Research Associate in the lab of Judith Mank, at UBC. My current work focuses on the evolution and genetic basis of the highly variable color patterns of the Trinidadian Guppy.
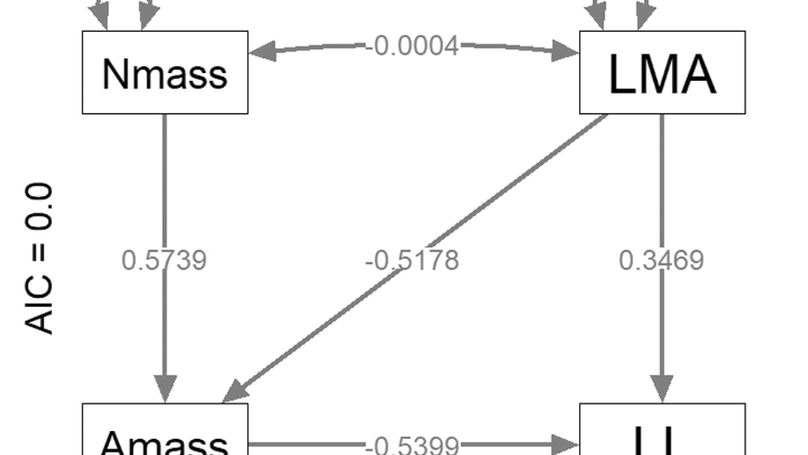
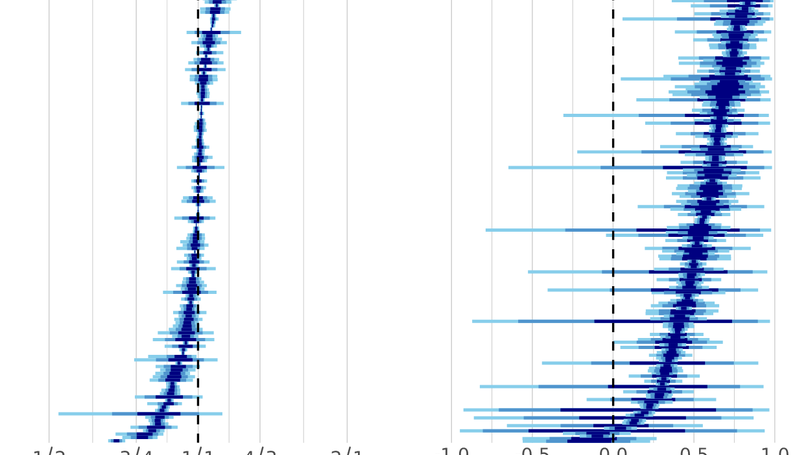
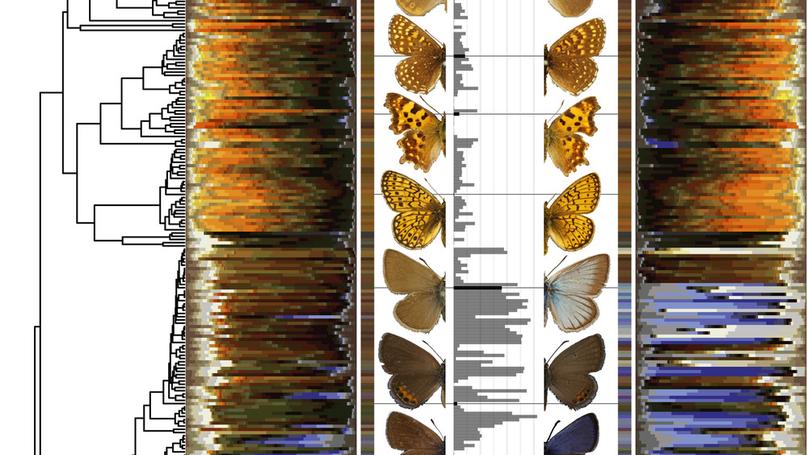
Previously, I worked in the lab of Chris Wheat on butterfly coloration. Before that, I was a PhD student with Niclas Kolm studying the function and evolution of brain size. For my thesis I performed experiments on brain size in guppies, and performed comparative analyses on the same topic.
- Trait evolution
- Sexual selection and coloration
- Genomics
- Quantitive genetics
- Behavior and cognition
- Phenomics & artificial intelligence
-
PhD in Ethology, 2018
Stockholm University
-
MSc in Behavioral and Cognitive Neuroscience, 2013
University of Groningen
-
BSc in Life Science & Technology, 2010
University of Groningen
Featured Publications
Recent Publications